關于征求國家生態(tài)環(huán)境標準《水質(zhì) 半揮發(fā)性有機物的測定 氣相色譜-質(zhì)譜法(征求意見稿)》
意見的通知
為貫徹《中華人民共和國環(huán)境保護法》�����,規(guī)范生態(tài)環(huán)境監(jiān)測工作,我部編制了國家生態(tài)環(huán)境標準《水質(zhì) 半揮發(fā)性有機物的測定 氣相色譜-質(zhì)譜法》征求意見稿�。按照國家生態(tài)環(huán)境標準制修訂工作規(guī)則要求,現(xiàn)公開征求意見(可登錄我部網(wǎng)站“意見征集”欄目檢索查閱征求意見稿及編制說明)�。
各有關單位和個人均可提出意見和建議。請將意見建議書面反饋我部�����,并注明聯(lián)系人及聯(lián)系方式���,電子文檔同時發(fā)送至聯(lián)系人郵箱�。征求意見截止時間為2021年5月16日�。
聯(lián)系人:生態(tài)環(huán)境部生態(tài)環(huán)境監(jiān)測司曹宇
地址:北京市東城區(qū)東安門大街82號
郵編:100006
附件:1.水質(zhì) 半揮發(fā)性有機物的測定 氣相色譜-質(zhì)譜法(征求意見稿)
2.《水質(zhì) 半揮發(fā)性有機物的測定 氣相色譜-質(zhì)譜法(征求意見稿)》編制說明
生態(tài)環(huán)境部辦公廳
2021年4月20日
(此件社會公開)
水質(zhì) 半揮發(fā)性有機物的測定 氣相色譜-質(zhì)譜法
警告:實驗中使用的有機溶劑和標準物質(zhì)為有毒有害化學品,試劑配制和樣品前處理過程應在通風櫥內(nèi)進行���,操作時應按要求佩戴防護器具�,避免直接接觸皮膚和衣物�����。
1 適用范圍
本標準規(guī)定了測定水中半揮發(fā)性有機物的氣相色譜-質(zhì)譜法���。
本標準適用于地表水����、地下水�����、工業(yè)廢水和生活污水中 64 種半揮發(fā)性有機物的篩查鑒定和定量分析����,對于特定類別的化合物,應在此篩選基礎上選用專屬的分析方法測定���。
當取樣體積為 1000 ml�����,試樣體積為 1.0 ml�,采用全掃描方式測定時�,方法檢出限為 0.1μg/L——2 μg/L,測定下限為 0.4 μg/L——8 μg/L���,詳見附錄 A�����。
2 規(guī)范性引用文件
本標準引用了下列文件或其中的條款�����。凡是不注日期的引用文件����,其有效版本適用于本標準。
HJ/T 91 地表水和污水監(jiān)測技術規(guī)范
HJ 91.1 污水監(jiān)測技術規(guī)范
HJ 164 地下水環(huán)境監(jiān)測技術規(guī)范
HJ 493 水質(zhì) 樣品的保存和管理技術規(guī)定
3 方法原理
用二氯甲烷分別在 pH>11 和 pH<2 的條件下�����,萃取樣品中的半揮發(fā)性有機物����。萃取液經(jīng)脫水、濃縮和定容后��,經(jīng)氣相色譜-質(zhì)譜法(GC/MS)分離檢測����,根據(jù)保留時間和目標化合物的特征離子定性�,內(nèi)標法定量����。
4 試劑和材料
除非另有說明��,分析時均使用符合國家標準的分析純試劑����,實驗用水為新制備的蒸餾水或不含目標化合物的純水。
4.1 二氯甲烷(CH2Cl2):農(nóng)殘級�����。
4.2 硫酸:ρ(H2SO4)=1.84g/ml����,優(yōu)級純。
4.3 氫氧化鈉(NaOH):優(yōu)級純����。
4.4 無水硫酸鈉(Na2SO4)。
在 400 ℃下灼燒或烘烤4h���,冷卻后裝入具塞磨口玻璃瓶中密封����,于干燥器中保存。
4.5 硫酸溶液:體積分數(shù)為 50%�。
將硫酸(4.2)與水按 1:1 體積比混合。
4.6 氫氧化鈉溶液:c(NaOH)=10.0 mol/L��。
稱取 40g 氫氧化鈉(4.3)溶于水中���,定容至 100 ml����。
4.7 半揮發(fā)性有機物混合標準貯備液:ρ=1000 μg/ml����。
市售有證標準溶液,按照說明書要求保存����。
4.8 色譜進樣口檢查液:ρ=50.0 μg/ml。
含有 4,4’-DDT��、五氯苯酚和聯(lián)苯胺濃度均為 50 μg/ml 的混合溶液���,市售��。
4.9 內(nèi)標化合物標準貯備液:ρ=2000 μg/ml���。
1,4-二氯苯-d4���,萘-d8�,苊-d10,菲-d10��,?-d12���,苝-d12���,市售有證標準溶液,按照說明書要求保存���。亦可選用其他性質(zhì)相近的半揮發(fā)性有機物做內(nèi)標�,并可以根據(jù)保留時間的范圍和種類適當調(diào)整���。
4.10 酸性替代物貯備液:ρ=10000 μg/ml�。
2-氟苯酚�����,苯酚-d6,2-氯苯酚-d4�,1,2-二氯苯-d4,2,4,6-三溴苯酚�����,市售有證標準溶液���,按照說明書要求保存���。
4.11 堿性替代物貯備液:ρ=5000 μg/ml。
硝基苯-d5�,2-氟聯(lián)苯,對三聯(lián)苯-d14�,市售有證標準溶液,按照說明書要求保存�。
4.12 酸性替代物使用液:ρ=1000 μg/ml。
用二氯甲烷(4.1)稀釋酸性替代物貯備液(4.10)��,臨用現(xiàn)配��。
4.13 堿性替代物使用液:ρ=500 μg/ml。
用二氯甲烷(4.1)稀釋堿性替代物貯備液(4.11)��,臨用現(xiàn)配����。
4.14 十氟三苯基膦溶液(DFTPP):ρ=1000 μg/ml。
市售有證標準品�,一般以二氯甲烷為溶劑。
4.15 十氟三苯基膦使用液:ρ=50.0 μg/ml����。
用微量注射器移取 500 μl 十氟三苯基膦溶液(4.14)至 10 ml 容量瓶中��,用二氯甲烷(4.1)定容至標線���,混勻�����。
4.16 高純氦氣:純度≥99.999%����。
4.17 高純氮氣:純度≥99.999%��。
5 儀器和設備
5.1 氣相色譜-質(zhì)譜儀:具有電子轟擊(EI)離子源。
5.2 色譜柱:30 m×0.25 mm 的熔融石英毛細柱��,膜厚 0.25 µm(5%苯基-95%二甲基聚硅氧烷固定液)����,或其它等效毛細管色譜柱。
5.3 濃縮裝置:配有帶 1.00 ml 刻度線濃縮管的氮吹儀���,或其他同等性能的設備�����。
5.4 微量注射器:10 μl��、50 μl�����、100 μl����、250 μl�����、500 μl。
5.5 分液漏斗:2000 ml����,具聚四氟乙烯旋塞。
5.6 采樣瓶:1 L 具聚四氟乙烯內(nèi)襯蓋或鋁箔包裹瓶蓋的玻璃磨口棕色瓶�。
5.7 250 ml 具塞錐形瓶。
5.8 一般實驗室常用儀器和設備��。
6 樣品
6.1 樣品采集
按照 HJ/T 91�����、HJ 91.1 和 HJ 164 的相關規(guī)定進行樣品的采集����,將樣品采集到采樣瓶(5.6)
中�,每批樣品應至少采集 1 個全程序空白樣品,用同批次實驗用水在現(xiàn)場裝滿采樣瓶(5.6)��,采樣結(jié)束后與樣品一起帶回實驗室����。
6.2 樣品保存
將采集好的樣品立即置于 4 ℃以下避光冷藏保存,于 7 d 內(nèi)完成萃取����。萃取后的濃縮液應在 40 d 內(nèi)分析完畢���。
6.3 試樣的制備
取 1000 ml 均勻樣品于分液漏斗(5.5)中,加入 20 µl 酸性替代物使用液(4.12)和 40µl 堿性替代物使用液(4.13)����,使最終試樣中替代物的濃度均為 20 μg/ml,混合均勻����。用氫氧化鈉溶液(4.6)調(diào)節(jié)樣品 pH>11,加入 30 ml 二氯甲烷(4.1)�,振搖萃取 10 min(萃取時注意周期性放氣釋放壓力),靜置分層���,收集有機相��,再重復以上萃取步驟兩次�����,合并 3次萃取液于 250 ml 具塞錐形瓶(5.7)中待用����。用硫酸溶液(4.5)調(diào)節(jié)水相 pH<2,分別用30 ml 二氯甲烷(4.1)萃取 3 次�����,有機相全部收集合并于錐形瓶中��。
將錐形瓶中的萃取液通過裝有無水硫酸鈉(4.4)的漏斗����,脫水后轉(zhuǎn)移至濃縮裝置(5.3)的濃縮管中。用少量二氯甲烷(4.1)多次淋洗無水硫酸鈉�,淋洗液合并于同一根濃縮管中。將濃縮管置于氮吹儀中����,在 40 ℃下用高純氮氣(4.17)將其濃縮至 0.5 ml——1 ml 之間。用二氯甲烷(4.1)定容至 1.0 ml��,加入 5 µl 內(nèi)標化合物標準貯備液(4.9)����,使試樣中內(nèi)標化合物的濃度為 10 μg/ml��,混勻待測���。
注 1:如遇成分復雜的樣品在萃取時發(fā)生乳化現(xiàn)象���,可采取機械手段完成兩相分離��,包括攪動�、離心�����、用玻璃棉過濾等方法破乳���,也可采用冷凍方法破乳���。
注 2:不同半揮發(fā)性有機化合物的定量分析時,可采用含有不同吸附劑的層析柱進行凈化����。不同目標化合物推薦使用的凈化方法見附錄E。
6.4 空白試樣的制備
用實驗用水代替樣品��,按照和試樣的制備(6.3)相同的步驟進行實驗室空白試樣的制備��。
7 分析步驟
7.1 儀器參考條件
7.1.1 氣相色譜參考條件
進樣口溫度:280 ℃��;不分流進樣;載氣:高純氦氣(4.16)�;進樣量:1.0 µl;柱流量:1 ml/min����;
初始溫度:40 ℃,保持 4 min��,以 8 ℃/min 的速率升溫至 300 ℃�����,一直保持到最后一個目標化合物苯并[g,h,i]苝出峰后�����。
7.1.2 質(zhì)譜參考條件
電子轟擊源(EI)����;離子源溫度:180 ℃;傳輸線溫度:280 ℃����;離子化能量:70 eV�;質(zhì)譜掃描范圍:35 amu——500 amu�����;數(shù)據(jù)采集方式:全掃描(Scan)模式或選擇離子掃描(SIM)��。
7.1.3 質(zhì)譜性能檢查
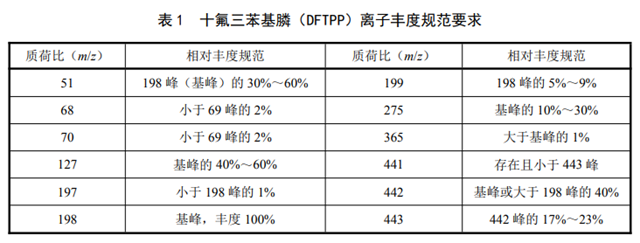
每次分析前���,應進行質(zhì)譜自動調(diào)諧,再將氣相色譜和質(zhì)譜儀設定至分析方法要求的儀器條件��,并處于待機狀態(tài)�,通過氣相色譜進樣直接注入 1.0 µl 十氟三苯基膦使用液(4.15),對整個系統(tǒng)進行檢查����,每運行 12 h 檢查一次,得到十氟三苯基膦質(zhì)譜圖���,其質(zhì)量碎片的離子的豐度應全部符合表 1 中的要求�。否則須清洗質(zhì)譜儀離子源�。
7.2 校準
7.2.1 標準曲線的建立
取 5 個 5 ml 容量瓶,預先加入 2 ml 二氯甲烷(4.1)����,分別取適量的半揮發(fā)性有機物混合標準貯備液(4.7)�、酸性替代物使用液(4.12)�、堿性替代物使用液(4.13)和內(nèi)標化合物標準貯備液(4.9),用二氯甲烷(4.1)定容后混勻�����,配制成至少 5 個濃度點的標準系列���。半揮發(fā)性有機物和替代物的質(zhì)量濃度分別為 5.0 µg/ml��,20.0 µg/ml�,50.0 µg/ml���,80.0 µg/ml���,100 µg/ml(此為參考濃度),內(nèi)標化合物的質(zhì)量濃度均為 10.0 µg/ml��。也可根據(jù)儀器靈敏度或樣品中目標化合物濃度配制成其他氣相色譜-質(zhì)譜儀合適的濃度水平校準系列�����。
按照儀器參考條件(7.1),從低濃度到高濃度依次進樣分析�,得到不同目標化合物質(zhì)譜圖。
7.2.2 平均相對響應因子的計算
標準系列第 i 點的目標化合物(或替代物)的相對響應因子���,按照公式(1)進行計算:

式中:RRFi——標準系列第 i 點目標化合物(或替代物)的相對響應因子;
Ai——標準系列第 i 點目標化合物(或替代物)的定量離子的響應值����;
AISi——標準系列第 i 點目標化合物(或替代物)的相對應內(nèi)標定量離子的響應值;
ρIS——標準系列內(nèi)標的質(zhì)量濃度��,μg/ml�;
ρi——標準系列第 i 點目標化合物(或替代物)的質(zhì)量濃度,μg/ml�����。
目標化合物或替代物的平均相對響應因子���,按照公式(2)進行計算:

7.3 試樣的測定
按照與標準曲線的建立(7.2.1)相同的儀器條件進行試樣(6.3)的測定����。
7.4 空白試驗
按照與試樣的測定(7.3)相同的儀器條件進行實驗室空白試樣(6.4)的測定���。
8 結(jié)果計算與表示
8.1 定性分析
通過樣品目標化合物與標準系列中的目標化合物的保留時間�、質(zhì)譜圖、碎片離子質(zhì)荷比及其豐度等信息比較���,對目標化合物進行定性分析�。應多次分析標準溶液得到目標化合物的保留時間均值���,以平均保留時間±3%的標準偏差為保留時間窗口����,樣品中目標化合物的保留時間應在其范圍內(nèi)�����。目標化合物標準質(zhì)譜圖中相對豐度高于 30%的所有離子應在樣品質(zhì)譜圖中存在����,樣品質(zhì)譜圖和標準質(zhì)譜圖中上述特征離子的相對豐度偏差應在±30%之內(nèi)。一些特殊的離子如分子離子峰��,即使其相對豐度低于 30%��,也應該作為判別化合物的依據(jù)���。
對沒有標準物質(zhì)或純品的半揮發(fā)性有機物�,可通過獲得的全掃描質(zhì)譜圖與 NIST 標準譜庫譜圖檢索進行定性。(1)分子離子峰應出現(xiàn)在樣品中��;(2)標準質(zhì)譜圖中相對豐度高于30%的特征離子應在樣品質(zhì)譜圖中存在�;(3)譜庫檢索可信度至少大于 70%。定性結(jié)果僅適用于污染初步篩查和未知物初步定性�����,并在報告中給出結(jié)果的可信度���。
在本標準推薦的儀器參考條件下,目標化合物的總離子流色譜圖參見附錄 B����。
8.2 結(jié)果計算
在對目標化合物定性的基礎上,根據(jù)定量離子的峰面積�����,采用內(nèi)標法進行定量����。當樣品中目標化合物的定量離子有干擾時,可使用輔助離子定量。定量離子�、輔助離子參見附錄 C。
當目標化合物(或替代物)采用平均相對響應因子進行校準時����,樣品中目標化合物(或替代物)的質(zhì)量濃度(μg/L),按照公式(3)進行計算:

8.3 結(jié)果表示
測定結(jié)果小數(shù)點后位數(shù)的保留與方法檢出限一致����,最多保留 3 位有效數(shù)字。
9 準確度
9.1 精密度
6 家實驗室對含半揮發(fā)性有機物濃度為 5 μg/L��、15 μg/L 和 60 μg/L 的統(tǒng)一空白加標樣品進行了 6 次重復測定:實驗室內(nèi)相對標準偏差分別為 1.8%——18.9%����、0.1%——17.7%和 0.1%——17.6%;實驗室間相對標準偏差分別為 1.7%——39%���、0.5%——50.3%和 2.2%——40.9%�����;重復性限為:0.4 μg/L——1.1 μg/L���、0.2 μg/L——2.1 μg/L 和 2.1 μg/L——12.2 μg/L����;再現(xiàn)性限為:0.5 μg/L——4.7 μg/L�����、0.8 μg/L——10.5 μg/L 和 4.5 μg/L——54.3 μg/L�����。
方法精密度匯總數(shù)據(jù)參見附錄 D�。
9.2 正確度
6 家實驗室分別對地表水樣品和工業(yè)廢水樣品(加標濃度為 20 μg/L)進行了 6 次重復加標分析測定:加標回收率范圍為 43.5%——96.8%和 43.0%——97.1%��;實驗室內(nèi)對未檢出目標化合物的地表水和工業(yè)廢水樣品(加標濃度為 20 μg/L)進行了 6 次重復加標分析測定:加標回收率范圍分別為 14.3%——95.5%和 13.2%——96.9%�����。
方法正確度匯總數(shù)據(jù)參見附錄 D��。
10 質(zhì)量保證和質(zhì)量控制
10.1 儀器性能檢查
用色譜進樣口檢查液(4.8)來檢查氣相色譜儀注射入口的惰性��,滴滴涕(DDT)到滴滴伊(DDE)和滴滴滴(DDD)的降解率不應超過 15%���,滴滴涕的降解率按公式(4)進行計算�。如果 DDT 衰減過多或出現(xiàn)較差的色譜峰,則需要清洗或更換進樣口�,同時還應截取毛細管柱前段約5 cm。聯(lián)苯胺和五氯苯酚等極性化合物在進樣口易出現(xiàn)分解����,峰形出現(xiàn)拖尾分裂等現(xiàn)象,也應進行同樣的處理�。

10.2 空白試驗
每批樣品應至少采集 1 個全程序空白,每 20 個樣品或每批次(≤20 個/批)至少做 1個實驗室空白���,測定結(jié)果中目標化合物濃度不應超過方法檢出限���。
10.3 校準
初始標準系列中目標化合物相對響應因子的相對標準偏差應不大于 30%。
每24 h分析1次標準系列中間濃度點溶液��,其測定值和標準值的相對誤差應在±30%以內(nèi)�。
10.4 平行樣
每 20 個樣品或每批次(≤20個/批)至少應分析1個平行樣,濃度水平在測定下限以上的平行樣測定結(jié)果的相對偏差應小于40%����。
10.5 基體加標
每 20 個樣品或每批(≤20個/批)至少做1個基體加標樣,加標濃度為原樣品濃度的1——5 倍或曲線中間濃度點���。目標化合物加標回收率的控制指標參見附錄D表D.2�����。
10.6 替代物回收率
實驗室應建立替代物加標回收率控制圖�����,按同一批樣品(20至30個樣品)進行統(tǒng)計��,剔除離群值����,計算替代物的平均回收率及相對標準偏差,替代物回收率應控制在平均值加減3 倍相對標準偏差范圍內(nèi)��。參見附錄D表D.4的替代物加標回收率的控制指標�����。
11 廢物處置
實驗中產(chǎn)生的廢液和其它廢棄物(包括檢測后的殘液)應分類收集�,集中保管���,并做好相應標識�����,依法委托有資質(zhì)的單位進行處理���。
12 注意事項
12.1 當分析高濃度樣品后連續(xù)分析低濃度樣品時���,可能由于過載而產(chǎn)生污染,需用溶劑清洗注射器���。分析完一個高濃度樣品后���,應考慮其對后續(xù)分析的樣品可能存在的干擾,需對儀器和分析環(huán)境進行檢查確認���,以確保分析系統(tǒng)不被污染�。
12.2 鄰苯二甲酸酯類化合物在實驗室普遍存在�����,樣品制備過程中應避免接觸塑料制品���,并檢查所有試劑空白�����,保證這類化合物在檢出限以下���。
12.3 六氯環(huán)戊二烯在氣相色譜進樣口易發(fā)生熱分解�����,與溶劑發(fā)生化學反應及光化學分解��;N-二甲基亞硝胺與溶劑共流出��,與二苯胺難分離�����,且在氣相色譜進樣口易發(fā)生熱分解�����,回收率不穩(wěn)定�。
12.4 當基體復雜有干擾時�����,針對相對應的目標化合物���,可以先采取相應的方法凈化��,然后再進行試樣的制備(6.3)�,但可能會對部分其他目標化合物的測定產(chǎn)生影響�,適用的凈化方法參見附錄E。